We have a new Progress in Retinal and Eye Research manuscript out in collaboration with my colleagues here at the Moran Eye Center.
Authors: Silke Becker, Zia L’Ecuyer, Bryan W Jones, Moussa A Zouache, Fiona S McDonnell, Frans Vinberg.
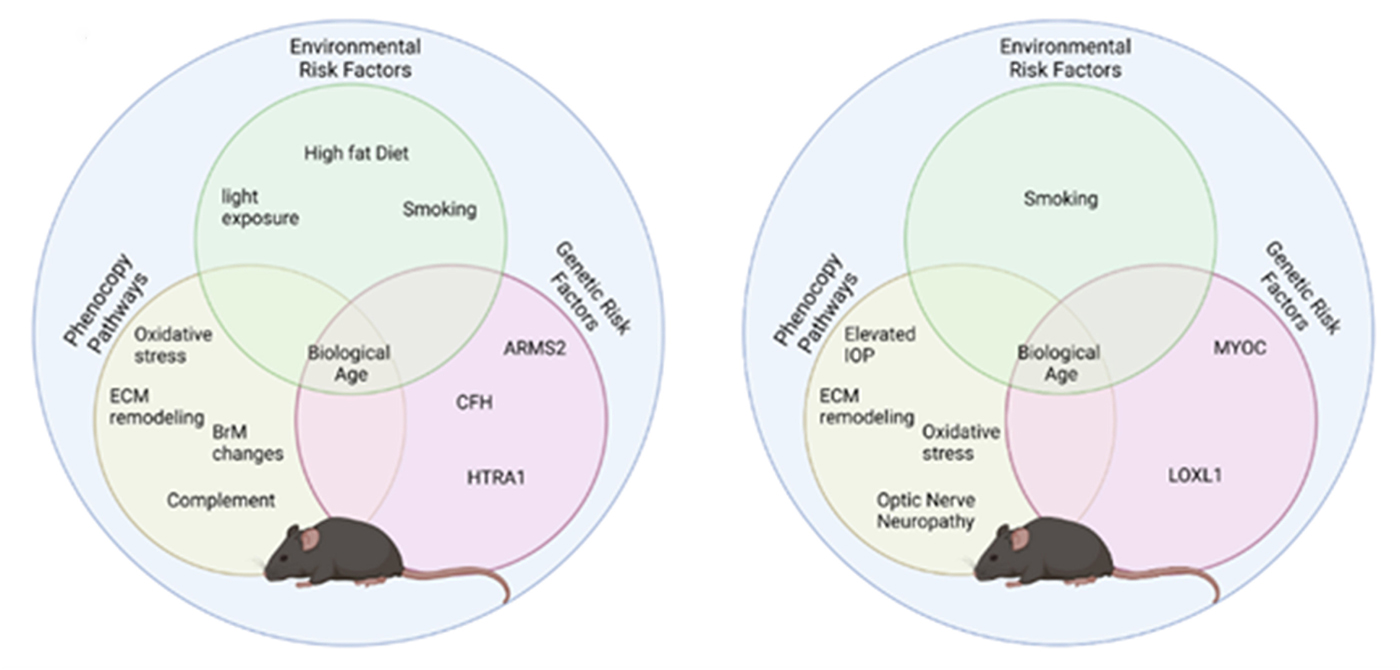
Abstract: Modeling complex eye diseases like age-related macular degeneration (AMD) and glaucoma poses significant challenges, since these conditions depend highly on age-related changes that occur over several decades, with many contributing factors remaining unknown. Although both diseases exhibit a relatively high heritability of >50%, a large proportion of individuals carrying AMD- or glaucoma-associated genetic risk variants will never develop these diseases. Furthermore, several environmental and lifestyle factors contribute to and modulate the pathogenesis and progression of AMD and glaucoma.
Several strategies replicate the impact of genetic risk variants, pathobiological pathways and environmental and lifestyle factors in AMD and glaucoma in mice and other species. In this review we will mostly discuss the most commonly available mouse models, which have and will likely continue to improve our understanding of the pathobiology of age-related eye diseases. Uncertainties persist whether small animal models can truly recapitulate disease progression and vision loss in patients, raising doubts regarding their usefulness when testing novel gene or drug therapies. We will elaborate on concerns that relate to shorter lifespan, body size and allometries, lack of macula and a true lamina cribrosa, as well as absence and sequence disparities of certain genes and differences in their chromosomal location in mice.
Since biological, rather than chronological, age likely predisposes an organism for both glaucoma and AMD, more rapidly aging organisms like small rodents may open up possibilities that will make research of these diseases more timely and financially feasible. On the other hand, due to the above-mentioned anatomical and physiological features, as well as pharmacokinetic and -dynamic differences small animal models are not ideal to study the natural progression of vision loss or the efficacy and safety of novel therapies. In this context, we will also discuss the advantages and pitfalls of alternative models that include larger species, such as non-human primates and rabbits, patient-derived retinal organoids, and human organ donor eyes.