We have a new manuscript in collaboration with the Gross Lab out of UAB in The FASEB Journal, (PubMed link here): NUDC is critical for rod photoreceptor function, maintenance, and survival. This manuscript is in collaboration with. Authors are: Mary Anne Garner, Meredith G. Hubbard, Evan R. Boitet, Seth T. Hubbard, Anushree Gade, Guoxin Ying, Bryan W. Jones, Wolfgang Baehr, Alecia K. Gross. The PDF is here.
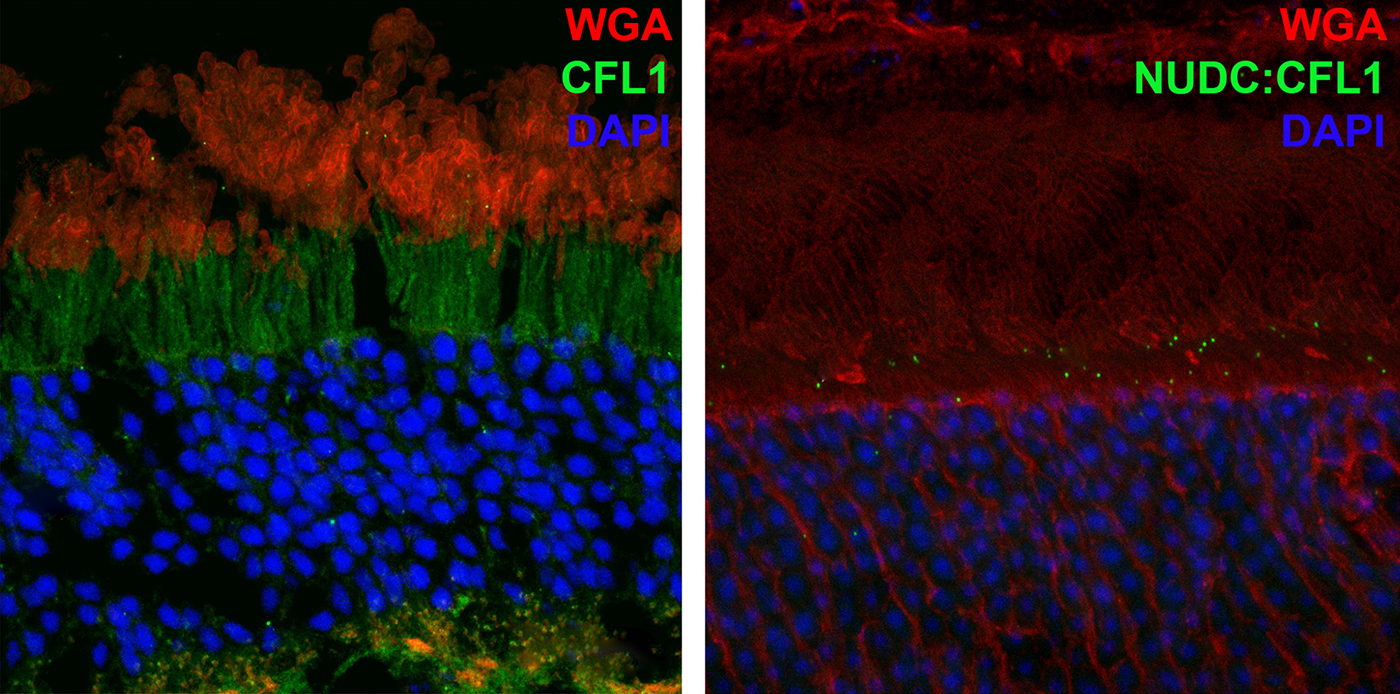
Abstract: NUDC (nuclear distribution protein C) is a mitotic protein involved in nuclear migration and cytokinesis across species. Considered a cytoplasmic dynein (henceforth dynein) cofactor, NUDC was shown to associate with the dynein motor complex during neuronal migration. NUDC is also expressed in postmitotic vertebrate rod photoreceptors where its function is unknown. Here, we examined the role of NUDC in postmitotic rod photoreceptors by studying the consequences of a conditional NUDC knockout in mouse rods (rNudC−/−). Loss of NUDC in rods led to complete photoreceptor cell death at 6 weeks of age. By 3 weeks of age, rNudC−/− function was diminished, and rhodopsin and mitochondria were mislocalized, consistent with dynein inhibition. Levels of outer segment proteins were reduced, but LIS1 (lissencephaly protein 1), a well-characterized dynein cofactor, was unaffected. Transmission electron microscopy revealed ultrastructural defects within the rods of rNudC−/− by 3 weeks of age. We investigated whether NUDC interacts with the actin modulator cofilin 1 (CFL1) and found that in rods, CFL1 is localized in close proximity to NUDC. In addition to its potential role in dynein trafficking within rods, loss of NUDC also resulted in increased levels of phosphorylated CFL1 (pCFL1), which would purportedly prevent depolymerization of actin. The absence of NUDC also induced an inflammatory response in Müller glia and microglia across the neural retina by 3 weeks of age. Taken together, our data illustrate the critical role of NUDC in actin cytoskeletal maintenance and dynein-mediated protein trafficking in a postmitotic rod photoreceptor.