This abstract was presented today, May 2th at the 2016 Association for Research in Vision and Opthalmology (ARVO) meetings in Seattle, Washington by Laurence Occelli, Bryan W. Jones, and Simon M. Petersen-Jones.
Posterboard #: D0250
Abstract Number: 2260 – D0250
Author Block: Laurence M. Occelli1 , Bryan W. Jones2 , Simon M. Petersen-Jones1
1 Small Animal Clinical Sciences, Michigan State University, East Lansing, Michigan, United States; 2 Ophthalmology, Moran Eye Center, University of Utah, Salt Lake City, Utah, United States
Disclosure Block:Laurence M. Occelli, None; Bryan W. Jones, None; Simon M. Petersen-Jones, None
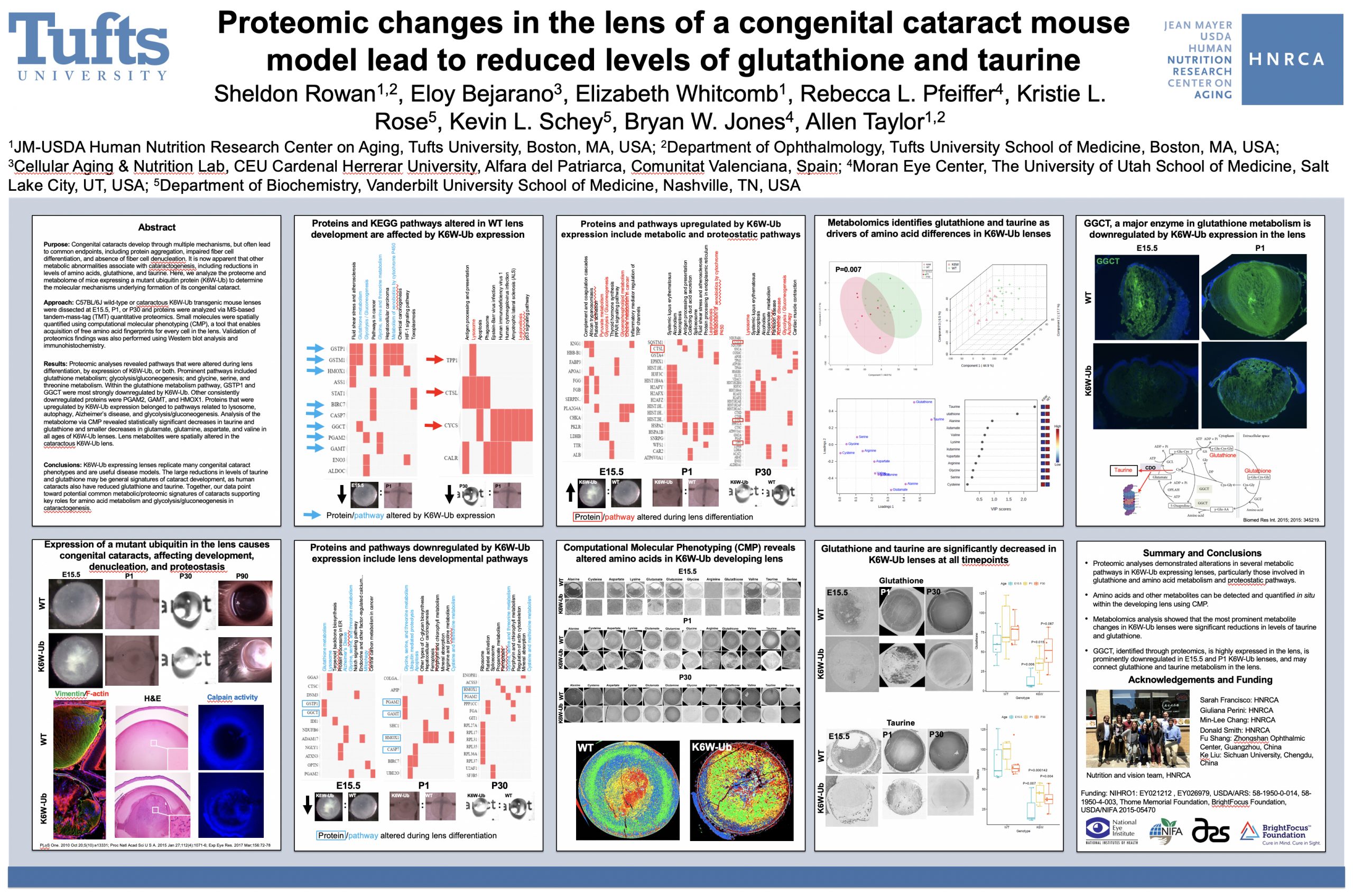
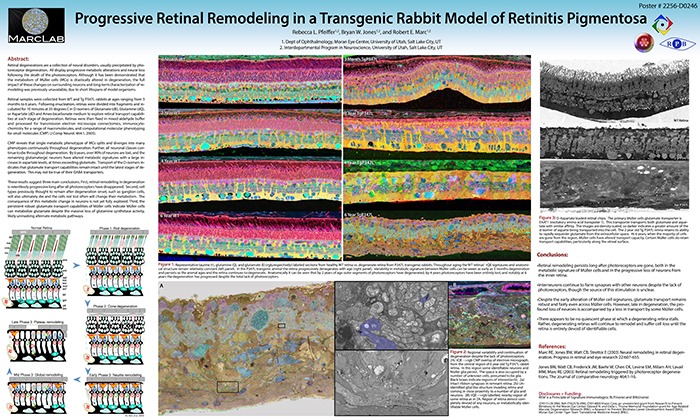
Purpose: CRX is a transcription factor essential for normal photoreceptor development and survival. The CrxRdy cat has a spontaneous mutation in Crx. Early disease stages in heterozygous cats (CrxRdy/+) mimics severe Leber’s congenital amaurosis. This study investigated the timing and extent of retinal remodeling in the late stages of retinal degeneration. This will help optimizing the best time for therapies such as retinal prosthesis or optogenetics before retinal rewiring and glial scar become too extensive.
Methods: CrxRdy/+ cats from 6 weeks to 10 years of age were investigated. Eyes were fixed in mixed aldehyde buffer and processed for immunocytochemistry for computational molecular phenotyping for macromolecules and small molecules (CMP) including GABA, glycine, glutamate, taurine, glutamine, aspartate, rhodopsin and red green opsin (J Comp Neurol. 464:1 2003). Samples from 5 retinal areas were collected: area centralis, mid- and far-superior as well as mid- and far-inferior regions.
Results: CMP revealed an absence of red green opsin and a decrease in rhodopsin expression with mislocalization to the photoreceptor inner segments (IS) and cell bodies as early as 6 weeks of age. Inner and outer photoreceptor segments (IS/OS) were present but short at 6 weeks of age. By 12 weeks of age, very few of the stunted OS remained and IS were very short. At that age, Müller cells had become activated initiating hypertrophy, and indicating cell stress. By 5 years of age, a Müller cell seal was clearly present disrupting the retinal lamination via glial columns. Migration of inner nuclear layer cells with inverted and everted cells was also observed from an early age as well as horizontal and amacrine cell sprouting. By 5 years of age, microneuromas formations had developed (Fig.1). Extreme thinning and remodeling was observed in the peripheral retina of older animals and retinal pigment epithelium was lost from the area centralis.
Conclusions: This study indicates that retinal degeneration in the CrxRdy/+ cat retina follows the 3 proposed phases of retinal remodeling. As early as 12 weeks of age, some glial reaction to photoreceptor death was observed followed by formation of a glial seal, rewiring and inner nuclear layer cells migration. Finally, microneuroma formation, severe retinal thinning and remodeling was developed.